

Хромозомните патологии са един от най-сериозните нарушения в клетките, поради удвояването на една от двойките хромозоми, възникват специални синдроми, които имат типични външни прояви и промени в метаболизма. Тризомията в една от двойките хромозоми (когато вместо две, има три хромозоми) се среща най-често в три варианта - синдром на Даун, който е най-разпространен и добре познат, а също и Патау и Едуардс. Те са получили имената си поради откритието и описанието им от учени..
Общи данни
Синдромът на Едуардс (или синдром на тризомия 18) е на второ място по отношение на появата на тризомия, след синдрома на Даун и се появява, когато 18-та двойка хромозоми не се счупи. Патологията се характеризира с различни нарушения в образуването на фетални системи и органи в утробата, има типични външни промени и неблагоприятни здравословни и житейски прогнози..
Важно еДецата попадат в категорията на хората с тежки увреждания, изискват специално отношение, грижи и грижи за своите родители и лекари..
 По отношение на разпространението, тази хромозомна аномалия се счита за доста честа, до 0,01-0,02% от децата, родени в света, имат тази хромозомна аномалия., и не зависи от определена раса или страна на пребиваване, е повсеместна. Според статистиката момичетата страдат 3-4 пъти по-често от момчетата, въпреки че няма добре обосновани научни данни за това явление. Точната причина за образуването на тризомия в плода не е определена, но са идентифицирани редица специфични рискови фактори, които увеличават вероятността за неговото развитие..
По отношение на разпространението, тази хромозомна аномалия се счита за доста честа, до 0,01-0,02% от децата, родени в света, имат тази хромозомна аномалия., и не зависи от определена раса или страна на пребиваване, е повсеместна. Според статистиката момичетата страдат 3-4 пъти по-често от момчетата, въпреки че няма добре обосновани научни данни за това явление. Точната причина за образуването на тризомия в плода не е определена, но са идентифицирани редица специфични рискови фактори, които увеличават вероятността за неговото развитие..
Наред с други хромозомни и генни разстройства, днес синдромът на Едуардс включва вродени и нелечими патологии, специални методи на грижа и медицинска подкрепа, рехабилитационни мерки, които позволяват да се подкрепи живота на децата по определен начин и да се изгради известен успех в неговото развитие - физически и психически. но поради драматичното разнообразие на нарушенията, които дава допълнителната хромозома, няма единни методи за лечение и грижи.
Историята на изследването на патологията
В медицинските трактати и художествена литература може да се намери единична информация за пациенти с този синдром в древни времена, но синдромът е описан подробно в началото на миналия век. Развитието на технологиите от онова време и преди това не позволяват подробно изследване на тази патология, така че причините за това не знаят. В допълнение, по-голямата част от децата, родени с този синдром, починаха в ранна възраст поради лошото развитие на медицината и навременната им помощ. Причината за синдрома - тризомията на 18-та двойка хромозоми е открита едва през 1960 г. от Д. Едуардс и на негово име е кръстена нова патология. Според медицинската статистика, появата на патология е средно 1 случай на 3000 концепции, но има много по-малко плодове, останали преди раждането. Това е така, защото подобна аномалия често в самото начало завършва с аборт или смърт в плода in utero. Но често при такива спонтанни аборти не се провеждат хромозомни изследвания..
Какво е тризомия
Говорейки от гледна точка на медицината, тризомията е вариант на хромозомни мутации, когато клетките на пациента нямат 46 хромозоми, събрани в хомоложни 23 двойки, чиято двойка определя половите разлики, но са открити 47 хромозоми..
Днес лекарите разграничават три вида тризомия, които принадлежат към нелетални мутации - синдром на Даун (21-та двойка страда), синдром на Патау (13-та двойка страда) и синдром на Едуардс (лезия се открива на 18-та двойка). Всички други варианти на тризомия първоначално са несъвместими с живота, такива ембриони веднага умират и бременността не се развива. Но раждането на деца с тези три тризомия винаги е проблем на тяхното здраве, физическо развитие и интелигентност, умствени функции..
Причини за възникване на синдром на Edwards
Подобно на другите две патологии, споменати по-горе, синдромът на Едуардс се нарича генетична патология, при която се открива допълнителна хромозома в автозомите (а не в половите хромозоми). Здравият човек има 46 хромозоми - 22 двойки - автозоми (без секс) и една двойка - пол, определящи дали това е момиче или момче. ДНК вериги са компактно опаковани в хромозоми, в които се записва цялата генетична информация за организма, започвайки от процеса на неговото зачеване до смъртта. Цялата програма на живота е написана на ДНК нишки..
При синдрома на Едуард тялото не страда от генни дефекти, а от хромозома, има дефект в разделението на зародишната клетка, който след това дава живота на детето, а допълнителната хромозома остава в него. В класическия вариант на формиране на синдрома на Едуард има дублиране на 18-та хромозома и едното момче и момиче могат да се родят с подобен синдром.. Има излишък на генетична информация във всички клетки на такова дете. Подобна излишък на ДНК води до редица нарушения, които водят до външни дефекти и проблеми в работата на системите и органите. Поради наличието на трета, допълнителна хромозома, се появи научно наименование - тризомия.
Видове синдром на Едуардс: как зависи тежестта на проявите
На базата на какъв вид хромозомни дефекти има в синдрома, има три вида потока:
- Пълна тризомия на 18-та двойка.
- Частична тризомия на 18-та двойка
- Мозаична форма на патология.
при пълна форма (също принадлежи към класическия синдром) се приема, че напълно във всички клетки има допълнителна хромозома. Този вариант е най-труден и се среща най-често, прогнозите с него са най-неблагоприятни..
 Частична тризомия се отнася до редки варианти на патология, възниква в 3% от случаите, като този проблем не съдържа пълната трета хромозома, а само неговото парче. Такъв дефект може да възникне в резултат на проблеми с клетъчното деление (особено техните хромозоми), което е изключително рядко. Може също да е вариант на факта, че допълнителната част от 18-та хромозома е прикрепена към отделни молекули ДНК, което води до тяхното удължаване и промяна. В тези клетки след това има две 18-и хромозоми и част от излишния материал. Тогава вроденият синдром няма да бъде толкова тежък, тъй като в клетките се появява допълнителна информация не на цялата информация, а само на части от нея.. Прогнозата за това състояние, макар и неблагоприятна, е много по-лесна за синдрома, отколкото за пълната форма..
Частична тризомия се отнася до редки варианти на патология, възниква в 3% от случаите, като този проблем не съдържа пълната трета хромозома, а само неговото парче. Такъв дефект може да възникне в резултат на проблеми с клетъчното деление (особено техните хромозоми), което е изключително рядко. Може също да е вариант на факта, че допълнителната част от 18-та хромозома е прикрепена към отделни молекули ДНК, което води до тяхното удължаване и промяна. В тези клетки след това има две 18-и хромозоми и част от излишния материал. Тогава вроденият синдром няма да бъде толкова тежък, тъй като в клетките се появява допълнителна информация не на цялата информация, а само на части от нея.. Прогнозата за това състояние, макар и неблагоприятна, е много по-лесна за синдрома, отколкото за пълната форма..
развитие мозаечна форма се появява при около 5-8% от децата с подобна аномалия. Но механизмите на този тип синдром са различни. Интересно е, че подобна форма се развива, след като сперматозоидът се слее с яйцеклетката и двете получени клетки първоначално имат нормален набор от хромозоми - 46ХХ или 46ХУ. Но още при първите разделения в образованата зигота се образува неизправност и една от клетките, придобити чрез тази допълнителна хромозома. В резултат на това ембрионът е придобил клетки с нормални хромозоми - 46 от тях, другата част има допълнителна хромозома - т.е. 47. Броят на клетките с допълнителна хромозома с такива проблеми няма да надвишава 50%.
Обърнете вниманиеКолкото по-голям е броят на дефектните клетки в ембриона и детето, толкова по-лоши са прогнозите и техният обем зависи от времето, в което е настъпил отказът по време на разделянето. Колкото по-късно се е случило, толкова по-малък е обемът на дефектните клетки, а броят на проблемите в развитието е намален..
Тази форма се нарича мозайка, защото нормалните клетки смесен с дефектен. Атипичните клетки с допълнителна хромозома са разпръснати произволно в цялото тяло и няма начин те да бъдат отстранени по какъвто и да е начин.. Тази форма е малко по-лесна, продължителността на живота е по-дълга..
Основата на патологията: каква е грешката на гените?
Само едно хромозома при дете има много сериозни и сериозни последствия за здравето и живота. Това се дължи на факта, че във всички клетки има универсална система за четене на информация и по време на клетъчното делене, удвояване на информацията, която е програмирана в ДНК молекулите. Ако има дефект на поне един ген - той вече засяга тялото под формата на заболяване или проблем, и ако има цяла част от допълнителната хромозома или дори цяла хромозома, има многобройни малформации и здравословни проблеми.
Хромозома 18 съдържа до 2.5% от цялата генетична информация за работата на тялото и нарушения, които се възпроизвеждат поради синдрома, са много значими. Екстра гените върху дефектната хромозома водят до синтеза на излишните протеини, което води до анормално развитие и функциониране на редица органи и системи на детето. При синдрома на Едуард, скелетът, някои зони на нервната система, урогениталната система и сърдечно-съдовата система са засегнати, те имат както аномалии в структурата, така и след като се развие плодът във функционирането..
Съответно, основната и единствена причина за този синдром е образуването на допълнителна хромозома в половата клетка на майката или бащата. При производството на зародишни клетки не се извършва обичайното разделение, последвано от удвояване на 23 двойки хромозоми до 46 броя (за всяко идентично копие в двойка), а само 23 броя. Ако в процеса на разделяне 18-та двойка по някаква причина не е правилно разделена, две хромозоми ще отидат на репродуктивната клетка едновременно и няма да има 23 броя в нея, а 24-d. Ако такава клетка води до нов живот (било то бащината сперма или майчиното яйце), това ще доведе до появата на дете със синдром на Едуардс..
Рискови фактори на синдром на Edwards
За да се генерират неправилно зародишните клетки на родителите и да се образува прекалено много хромозоми, са необходими различни негативни влияния, както отвън, така и от вътре в тялото.. Водещите от тях могат да бъдат разглеждани:
- възраст на майката и бащата по време на зачеването. Има реални доказателства за възрастовия фактор за вероятността от тризомия. С напредването на възрастта броят на анормалните клетки в репродуктивната система се увеличава, което е свързано с отслабване на контрола на организма върху процесите на разделяне. Рискът от раждане на деца с хромозомни патологии след 30 години се увеличава с 2-3 пъти в сравнение с младата възраст, а след 40 години шансовете се увеличават с 6-8 пъти повече. Зависимостта от възрастта на бащата не е толкова очевидна, но са възможни и хромозомни дефекти..
- имат лоши навици, особено ако е пристрастеност към никотин и алкохол. Обичайните интоксикации нарушават процесите на клетъчно делене, включително в гениталната област, което води до увеличаване на риска от дефектни сперматозоиди и яйца няколко пъти. Още по-опасно е употребата на наркотични вещества и токсикомания, те допълнително нарушават процесите на клетъчно делене..
- приемане на някои лекарства, повлияващи процесите на клетъчно делене, прекомерни дози и продължителност на приложение, неблагоприятни комбинации на лекарства по време на лечението, образувайки токсични концентрации от тях.
- соматични и генитални инфекции, които водят до поражение на репродуктивните органи и клетки, образуват хронично течение, засягат процесите на деление (цитомегалия, херпес, папилом и др.).
- професионални рискове, различни видове експозиция - йонизиращи, магнитни и много други. Те засягат процеса на клетъчно делене, оказват вредно въздействие върху ДНК, водят до образуването на свободни радикали в клетките и мутации.
Крайните причини и всички рискови фактори, които водят до образуването на клетки с тризомия, могат да бъдат изяснени като открития на молекулярната биология и генетика. Всички тези фактори само увеличават рисковете, но не казвайте, че такова дете ще се роди..
Не е склонна към образуване на тризомия в зародишните клетки, това, което се доказва от факта, че при раждане в семейството на едно дете със синдром на Едуард, шансовете за раждане на друго бебе се увеличават 200 пъти в сравнение със средното - от 0.04% до 2-3% или повече.
Характеристики на синдрома на Едуард в неонаталния период
Въпреки че днес е възможно да се диагностицира синдромът преди раждането, често се открива патология още при раждането. Новороденото с подобна хромозомна аномалия има типични и изразени малформации, тяхното присъствие може да помогне за поставяне на диагноза дори и без допълнителни изследвания на хромозомите - генетичен анализ (но все пак се прави, без тези данни диагнозата не е окончателна).
При раждане, идентифицирани:
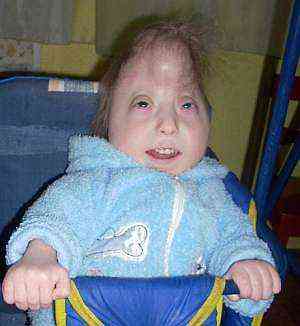 аномалии в структурата на формата на черепа. Скелетът с такъв синдром страда най-силно - това се проявява в промяната предимно във формата на черепа с образуването на долихоцефалия (рязко удължен, тесен череп - "кула"). Такава аномалия не е характерна само за тази патология, но показва съществуващите аномалии в развитието. Най-изразена промяна в съотношенията според измерванията в областта на париеталните кости и дължината от носа до тилната туберкула. По-тежката и тежка аномалия на черепа може да бъде микроцефалия, непропорционално малък размер на мозъчния череп по отношение на тялото..
аномалии в структурата на формата на черепа. Скелетът с такъв синдром страда най-силно - това се проявява в промяната предимно във формата на черепа с образуването на долихоцефалия (рязко удължен, тесен череп - "кула"). Такава аномалия не е характерна само за тази патология, но показва съществуващите аномалии в развитието. Най-изразена промяна в съотношенията според измерванията в областта на париеталните кости и дължината от носа до тилната туберкула. По-тежката и тежка аномалия на черепа може да бъде микроцефалия, непропорционално малък размер на мозъчния череп по отношение на тялото..- специфично модифицирана форма на ушната мида. Анормалната структура на ушите при новородени е силно изразена и се наблюдава при 95% от бебетата, особено при пълния тип на синдрома. Ушите са много по-ниски от обикновено, сравнително рязко понижени спрямо орбитите, хрущялите са изпъкнали и няма ясно оформени къдрици. Ушите на ухото и трагусът са в ранна детска възраст или са напълно отсъстващи, слуховият зъб рязко се стеснява и една четвърт от децата могат да отсъстват напълно..
- анормална структура на небето с долната челюст (образуване на микрогнатия). При новородените, когато се подозира синдромът на Edwards, няма пълно нарастване на неблагоприятните процеси в зоната на горната челюст, които образуват надлъжен процеп. Възможно е да има опции с пълно отцепване само на мекото небце или частично сливане, понякога с прехода към устните. При пълната форма на дефекта, неразкъсването може да бъде двустранно, горната устна е разделена от двете страни, което води до патологични цепнатини в челюстите и небцето, което пречи на детето напълно да затвори устата си. В случай на тежка патология е възможна комуникация между орофаринкса и носните кухини, дори ако устата е затворена. Честотата на такива аномалии достига 20-25% и по-висока..
- Възможно е да има и други проблеми. - готическо небе (прекомерно висок костен свод) и промени в структурата на долната челюст. Те са регистрирани в около 70% от децата. типичен микрогнатия - брадичката и долната челюст са силно изтеглени назад, което предпазва бебетата от пълно затваряне на устата и изтичане на слюнка от устата. При такава аномалия има затруднения с храненето, особено чрез нанасяне на гърдата..
- деформация на стъпалата с образуване на "разклащане на крака", характерните аномалии на дължината на пръстите на крайниците. Промяната в стъпалото е типична за 75% или повече деца на фона на този синдром, има промяна в положението на костите - глезен, пета или лопатка, което води до образуването на силно изпъкнали пети срещу отсъствието на крак арка. Тя може да се наведе напред, давайки на краката поглед на краката от люлеещ се стол. Откриват се и дисбаланси в дължината на пръстите на краката, особено на големите. Той е по-къс от втория пръст на крака, както се вижда с прилепнали пръсти.
- ръце в положение на огъване и сраствания между пръстите, специални дерматоглифични рисунки. aschistodactylia (този термин се отнася до сливането на пръстите) се открива в 45% от случаите, обикновено това са пръстите на краката, но ръцете могат да бъдат засегнати. В лека степен кожата създава мембрани, а костните прегради могат да се образуват в тежки. Може също така да бъде гъвкави аномалии на ръцете - положението на пръстите в аномалното, постоянно напрегнато положение, покриващо с малкия пръст и палеца всички останали, плътно притиснати към дланта. Такива нарушения са характерни за 90% от децата със синдрома..
- Наред с другите неща, с типична специална трисомия модели на пръстите и дланите със специална форма на кожни гънки. С развитието на този синдром, аномалии са типични за 60% от бебетата. Това са много чести дъги върху подложките, липсата на кожна гънка между екстремните и предпоследните фаланги, развитието на напречния жлеб - има термина "маймуна"..
- генитални малформации. При момчетата това е недоразвитие на пениса и скротума, при момичетата - увеличаване на размера на клитора. Аномалиите обикновено се комбинират с дефекти на пикочно-половата система, но очевидно останалите знаци не се виждат.
 аномалии в структурата на формата на черепа. Скелетът с такъв синдром страда най-силно - това се проявява в промяната предимно във формата на черепа с образуването на долихоцефалия (рязко удължен, тесен череп - "кула"). Такава аномалия не е характерна само за тази патология, но показва съществуващите аномалии в развитието. Най-изразена промяна в съотношенията според измерванията в областта на париеталните кости и дължината от носа до тилната туберкула. По-тежката и тежка аномалия на черепа може да бъде микроцефалия, непропорционално малък размер на мозъчния череп по отношение на тялото..
аномалии в структурата на формата на черепа. Скелетът с такъв синдром страда най-силно - това се проявява в промяната предимно във формата на черепа с образуването на долихоцефалия (рязко удължен, тесен череп - "кула"). Такава аномалия не е характерна само за тази патология, но показва съществуващите аномалии в развитието. Най-изразена промяна в съотношенията според измерванията в областта на париеталните кости и дължината от носа до тилната туберкула. По-тежката и тежка аномалия на черепа може да бъде микроцефалия, непропорционално малък размер на мозъчния череп по отношение на тялото..В допълнение към тези прояви, има и по-малки признаци и аномалии в развитието на децата, които могат в предварителната диагноза на синдрома. Важно е да се отбележи, че един от изброените симптоми и дори двама все още не говорят за диагнозата, има многобройни дефекти в синдрома и имат различни комбинации..
Важно еПовечето от описаните прояви могат да се появят като единични варианти и не потвърждават диагнозата, те могат да бъдат изолиран вариант на аномалии по време на бременност или проява на други патологии. Диагностицирането само на външни дефекти не може да се направи.!
Освен това, на фона на непълни или мозаични типове на синдрома на външни признаци, може да има малко една или две, докато има сериозни дефекти на вътрешните органи с чести аномалии в хромозомите. Именно те определят бъдещите прогнози за живота и хода на синдрома..
Развитие на детето: особености на синдрома на Едуардс
Единствено появата с появата на светлината и проявите на малформации в развитието, аномалията на Едуард не е ограничена, тъй като тя расте и се развива, прояви на нови и по-тежки отклонения са възможни. Първият от тях може да бъде идентифициран в рамките на няколко седмици от момента на раждането. Често за първи път е възможно да се подозира диагноза за мозаична и непълна форма на патология, ако външните дефекти са малко или никакви.. Гореописаните дефекти и външни промени стават по-остри и по-изразени, докато растат, постепенно се добавят нови проблеми и аномалии, които не се забелязват при раждането. Така че, тук си струва:
- Остро и ясно изразено изоставане в нивото на физическото развитие. При раждането масата на тези деца не надвишава 2-2,5 кг, а причината за това са хромозомните дефекти, които не позволяват системите и органите да се развият напълно. С напредването на развитието растежът и телесното тегло варират значително от месец на месец, а обхватът на гърдите също е слаб. Размерите на главите могат да достигнат или над нормата, което може да покаже натрупване на излишната течност в черепната кухина, образувайки хидроцефалия.
- Проблеми с крака. Тъй като те растат, се образува клисура поради аномалии в структурата на костите и сухожилията, мускулния хипертонус. Страдание и контрол върху развитието на стъпалото и тонуса на нервната система. Това е застрашено от факта, че децата започват да ходят късно или не могат да го направят изобщо поради общото тежко състояние. Външно, клисура се проявява в тежка деформация на крака и необичайна инсталация в покой.
- Нарушение на мускулния тонус. При раждането хипертоничността е типична за ръцете, оформяйки тяхната флексорна позиция, но докато се развива, тя се разпространява към съседни мускулни групи, водещи до изкуствени пози. Но по-често мускулния тонус се намалява, децата са бавни и атонични, крайниците се претеглят с камшици. Може да има и състояния на дистония с рязко свиване на отделните мускулни групи на фона на хипотонията на други. В този случай ръцете могат да бъдат огънати, а краката могат да се прегънат. Това води до нарушена координация и хаотични движения, изкълчвания и необичайно положение на крайниците..
- Атипични реакции на нервната система, емоционални проблеми. Някои части на мозъка при деца със синдром на Едуард са слабо развити, поради което се формират проблеми на емоционалност и инхибиране, неадекватни реакции към околната среда. Обикновено се свързва с мазолестото тяло и мозъчните хипоплазии, които също се образуват умствена изостаналост. Външно това се проявява в отсъстващия поглед, липсата на емоция и невъзможността за установяване на контакт, липсата на проследяващи обекти. Децата не реагират на звуци, дължащи се на проблеми с ушите и нервната система, такива аномалии се откриват в първите месеци от живота..
Като цяло, поради тежки и комбинирани лезии, децата не живеят до пълнолетна възраст.. Някои от тях с мозаична форма могат да достигнат до възрастта на подрастващите с дълбока степен на ибетичност и външни прояви на малформации..
Диагностични техники за синдром на Edwards
Днес има три вида диагностичен синдром, които са фундаментално различни един от друг.. Предвид факта, че патологията е нелечима и смъртоносна в ранна възраст, важно е да се обърне внимание на пренаталната диагностика, която не позволява раждането на деца с тези аномалии.. Консултации на генетици в семейства, където има случаи на хромозомни аномалии, също ще бъдат полезни.. Можете да похарчите:
- Диагностика на зародишните клетки преди зачеването. За съжаление, този метод е приложим все още е много ограничен и само в IVF протоколи в предварителното проучване на зародишните клетки. Ако това е естествено схващане, лекарите могат да предскажат по-високи рискове за раждането на такива деца, но това не е нищо повече от прогнози. Невъзможно е да се знае със сигурност дали такова дете ще се роди или не. Семейната анамнеза се изследва, като се вземат предвид всички съществуващи заболявания, оценяват се рисковите фактори и се извършва генетичен анализ за двамата родители, като се използва сложно оборудване и тестове. Чрез определяне на кариотипа на родителите могат да се направят някои заключения..
- Идентифициране на синдрома по време на бременността. С развитието на плода, има няколко метода за пряко или косвено потвърждение на синдрома на Едуардс, първоначално за тези цели, ултразвуков скрининг и кръвни тестове се използват при проучване на специфични показатели, и при висок риск се извършват инвазивни процедури - амниоцентеза или кордоцентеза и биопсия на хорионна вълна. Провеждайте скрининг в строго определени периоди, когато промяната в данните от ултразвука и кръвните параметри ще бъде най-значима.
Получавайки директно клетките на плода или неговата кръв ви позволява да провеждате изследване на неговите хромозоми и да потвърждавате или отричате синдрома на Едуардс. Това помага при вземането на решение дали да се удължи или прекрати бременността по медицински причини..
- Потвърждаване на синдрома след раждането. Извършва се според резултатите от външни данни в комбинация с редица генетични анализи, потвърждаващи наличието на тризомия на 18-та двойка хромозоми. Освен това е важно да се оцени състоянието на детето и да се направят прогнози за неговия живот и здравословно състояние, за да се разбере колко помощ му е необходима, за да оцелее.. Без медицинска грижа за този синдром децата могат бързо да умрат в областта на раждането през следващите седмици.. УЗИ и ехокардиография, рентгенография и ЕЕГ, цяла поредица от изследвания на кръвта и урината, допълнителни изследвания до ЯМР и КТ.
Лечение и прогноза за синдром на Edwards
За съжаление, този синдром е неизлечим до момента и лекарите могат само да помогнат на детето да се справи по-добре с различни проблеми чрез хирургични интервенции и консервативна терапия.. Обикновено децата живеят до няколко месеца или година, но само с частична или мозаична форма могат да живеят по-дълго. Смъртта настъпва от многобройни малформации и аномалии в работата на органите, както и от свързването на свързани усложнения. Радикално коригиране на дефектите на хромозомите в клетките на детското тяло на настоящия етап от развитието на медицината е невъзможно.
Альона Парецкая, педиатър, лекар