

Мукополизахаридозата е цяла гама от генетично определени (обусловени) патологии, които се появяват в случай на нарушен метаболизъм на кисели мукополизахариди (гликозаминогликани).
Най-често с такова нарушение се засяга скелетът и физическото развитие се забавя, а някои видове патология се проявяват в изостаналост от страна на интелектуалната сфера. В допълнение, има неуспехи в сърдечно-съдовата, стомашно-чревната и нервната системи, оптичните апарати и кожата.
Лечението на мукополизахаридоза е симптоматично, прогнозата е като цяло неблагоприятна - уврежданията напредват, тъй като глюкозаминогликаните се акумулират. Пациентите умират достатъчно рано.
Общи данни
Мукополизахаридозата е изследвана сравнително неотдавна - през 20-ти век. Първоначално се разглеждаше като единна патология, след което, поради разнообразието, те започнаха да различават неговите видове. Обединяващият фактор на цялото мукополизахаридоза е, че киселите мукополизахариди се натрупват в органи и тъкани, а причината за това нарушение е по-ниската степен на някои клетъчни ензими, която се инкорпорира в генотипа и се наследява.
Обърнете вниманиеОртопедите лекуват пациенти с мукополизахаридоза, но изискват и клинични усилия от кардиолози, офталмолози, невролози, отоларинголози и някои други специалисти..
Мукополизахаридоза тип IH
Мукополизахаридоза тип IH се нарича също синдром на Hurler. Диагностициран е при 1 новородено за 20-25 хиляди.
Първите признаци на заболяването започват да се появяват през първата година от живота на детето, но пълна клинична картина може да се развие само за 2 години..
Най-типичните симптоми на заболяването са:
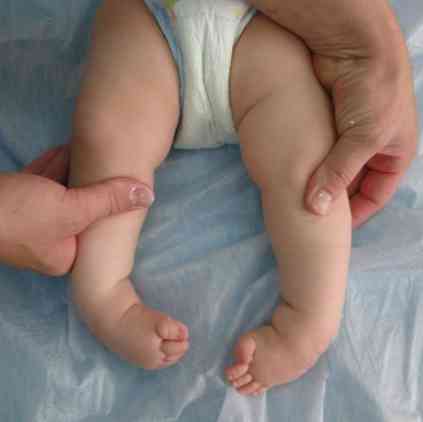 груби очертания на лицето - много типично за тази патология;
груби очертания на лицето - много типично за тази патология;- деформация на черепа - става като килов лодка. Това състояние се нарича саракафалия;
- дишането се извършва през устата, тъй като носното дишане се влошава поради увеличаване на аденоидите, както и интраутеринни аномалии в развитието на лицевия череп;
- деформация на крайниците и някои други фрагменти на скелета;
- забавяне на растежа;
- изкривяване на гръбначния стълб (симптом "котешки гръб" в седнало положение);
- скъсяване на шията;
- атипично високо разположение на лопатките;
- изпъкнали предни долни ребра;
- увеличаване на ширината на четките - докато те приличат на нокът с нокът;
- развитие на контрактури (намаляване на обхвата на движение) с постепенно оттегляне на различни стави в патологичния процес. Първо са засегнати лакътните и раменните стави, след което процесът се простира до коленните, тазобедрените и глезените стави.
 груби очертания на лицето - много типично за тази патология;
груби очертания на лицето - много типично за тази патология;Синдромът на Hurler се характеризира със специфична походка - на пръсти с извити крака. Такава характеристика възниква поради ограничаването на движенията, дължащи се на контрактури..
Физическият преглед показва следното:
- коремът е разширен, предната му стена при такива пациенти е отслабена, затова често се развиват пъпна и ингвинална херния;
- черния дроб и далака увеличени;
- при мъжете често се открива хидроцеле - течаща капка;
- в някои случаи се откриват нарушения на координацията на движенията;
- разкрива прекомерен растеж на пухкава коса.
Когато инструменталният преглед се определя от следното:
- на електрокардиограмата - признаци на дифузно (широко разпространено) увреждане на сърдечния мускул;
- увреждане на зрението и слуха;
- офталмоскопия разкрива помътняване и разширяване на роговицата.
Най-характерни са промените в състоянието по време на рентгеновото изследване:
- Рентгенова снимка на гръбначния стълб - позволява да се идентифицира характерната деформация на гръбначния стълб. В този случай прешлените имат формата на куб, краищата им са заоблени. Също така белязана кифоза - изкривяване на гръбначния стълб напред;
- рентгенография на гръдния кош - забележимо удебеляване на предните фрагменти на ребрата с едновременно изтъняване на задните им участъци, скъсяване и деформация. В същото време ребрата придобиват специфичен външен вид и стават подобни на лопата или сабя. Отбелязват се също намаление и известно изместване на главите на раменната кост;
- рентгенография на таза - разкрива стесняване на тазовия пръстен, ацетабуларните кухини (онези в които са "основата" на образуването на тазобедрената става) са изкривени, а главите на бедрените кости са намалени;
- Рентгенова снимка на тубуларни кости - изтъняване на кортикалния (повърхностен) слой на костите и известно разширяване на медуларния канал;
- Рентгенова снимка на костите на ръката - помага за откриване на недоразвитост на дисталните (нокътни) фаланги на пръстите, скъсяване и разширяване на средните и проксималните (тези по-близки до дланите) фаланги, както и метакарпалните кости;
- Рентгенография на черепа - ясно недоразвити кости на лицевия череп, краниостеноза (стесняване на черепа на някои места) и макроцефалия (увеличаване на черепа).
При пациенти с диагноза мукополизахаридоза тип IH се установяват следните нарушения, които могат да се разглеждат като отделни заболявания:
- ретинална пигментна дистрофия - нарушение на неговото развитие;
- глаукома - повишаване на вътреочното налягане;
- конгестия във фундуса;
- пареза - нарушение на движението;
- парализа - пълно нарушаване на двигателната активност в определени области на опорно-двигателния апарат.
С течение на времето пациентите с диагноза Hurler развиват и необратимо прогресират умствената изостаналост.
Мукополизахаридоза тип I-S
Друго име за мукополизахаридоза тип I-S е заболяването на заболяването. Заболяването е описано едва в средата на 20-ти век, въпреки че в практиката на клиницистите се срещат пациенти с характерни контрактури на пръстите на ръцете, двигателни нарушения при големи стави и специфични особености на лицето. Симптомите могат да се комбинират със симптоми на синдром на Hurler.
Този вид мукополизахаридоза се появява на по-късна възраст, отколкото при мукополизахаридоза тип IH, и неговият курс е по-благоприятен..
Характерна особеност на мукополизахаридоза тип I-S е, че до 3-6-годишна възраст развитието на децата съответства на възрастовата норма. Освен това започват да се появяват следните знаци:
- свиващи контрактури на пръстите;
- ограничаване на разширяването в големи и средни стави, а именно лакът, рамо и китка.
Нарушаването на амплитудата на движенията от страна на долните крайници е по-слабо изразено, отколкото при други видове описана патология.
Пълната клинична картина на този вид мукополизахаридоза е установена в началото на юношеството.
Физиологичното изследване (изследване) отбелязва следното:
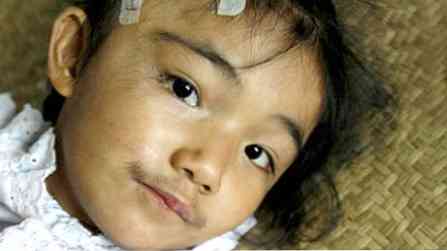 пациентите не са високи, но имат силна постройка (набит);
пациентите не са високи, но имат силна постройка (набит);- характеристиките са доста груби;
- добре развита скелетна мускулатура;
- се наблюдава хипертрихоза (повишена телесна коса);
- кожата на пръстите на ръцете (по-често) и краката (по-рядко) е удебелена и опъната;
- размерът на черния дроб и далака, като правило, не се променя.
 пациентите не са високи, но имат силна постройка (набит);
пациентите не са високи, но имат силна постройка (набит);В допълнение, тези пациенти са идентифицирани нарушения, които могат да се разглеждат като отделна болест:
- ингвинална или пъпна херния;
- синдром на карпалния тунел - тя се развива поради изразена компресия на средния нерв. Той се проявява чрез атрофия (хипоплазия) на тенарните мускули (повдигане в областта на палеца) и парестезии (нарушение на чувствителността) в областта на 3, 4 и 5 пръста;
- аортна стеноза - стесняване на аортата;
- аортна недостатъчност;
- ретинална пигментна дистрофия;
- глаукома;
- замъгляване на роговицата.
Интелигентността при пациенти с диагноза мукополизахаридоза тип I-S остава нормална, за разлика от пациенти с мукополизахаридоза тип IH.
От инструменталните методи най-информативна е радиографията. Той идентифицира промени, които са подобни на промените в мукополизахаридоза тип IH, но те са по-слабо изразени..
Мукополизахаридоза тип II
Този тип мукополизахаридоза също се нарича болест на Хънтър. Патологията най-често засяга момчета на възраст 2-3 години.
Според клиничните прояви, заболяването е подобно на мукополизахаридоза тип IH - докато има:
- skarotsefaliya;
- груби черти;
- нисък тон на гласа;
- затруднено дишане - поради деформация на костите на лицевия скелет.
При по-нататъшно развитие на заболяването се присъединяват следните признаци:
- намалена интелигентност;
- нарушена е координацията на движенията;
- Настроението при такъв пациент е нестабилно - отбеляза се така наречената емоционална лабилност - от пристъпи на смях до плач. Такива промени в настроението могат да възникнат по всяко време..
С прогресирането на мукополизахаридоза тип II се появява и увеличава неоправданата агресивност.
За разлика от мукополизахаридоза тип IH, нарушения на гръбначния стълб под формата на кифоза не са характерни, симптом на "котка обратно" не се наблюдава.
Физическият преглед определя следното:
- открива се слабо увеличение на далака и черния дроб;
- върху кожата на гърба се определят специфични възли.
Когато инструменталният преглед разкри:
- непрозрачност на роговицата, която напредва;
- увеличаване на загубата на слуха.
От всички инструментални методи за диагностика, най-информативна е рентгенографията на различни фрагменти на опорно-двигателния апарат - черепа, рудата, таза и т.н. В същото време, радиологичните признаци са същите, както при мукополизахаридоза тип I-S.
Пациентите с такава диагноза са предразположени към развитие на заболявания на дихателната система:
- трахеит;
- бронхит;
- пневмония.
Трябва да се отбележи, че мукополизахаридозата тип II може да се развие под формата на две възможности:
- неблагоприятна (вариант А);
- благоприятно (вариант Б).
 Неблагоприятният вариант се проявява с ярка клинична картина, като същевременно се забелязват съществени нарушения на интелигентността. Смъртните случаи могат да възникнат по време на юношеството.
Неблагоприятният вариант се проявява с ярка клинична картина, като същевременно се забелязват съществени нарушения на интелигентността. Смъртните случаи могат да възникнат по време на юношеството.
Симптомите с благоприятен вариант не са изразени, горните нарушения могат да бъдат придружени от лека умствена изостаналост. Такива пациенти, като правило, живеят до 30 години, в някои случаи - и повече.
Мукополизахаридоза тип III
Мукополизахаридоза тип III е описана от американски педиатър Sanfillipo, така че патологията е получила друго име - болест на Sanfillipo.
Заболяването се диагностицира при 1 дете на 100-200 хиляди новородени..
Децата с мукополизахаридоза тип III в първите години от живота си се развиват според възрастовите норми и могат да се наблюдават някои прояви на патология - по-специално затруднено преглъщане и неудобна походка..
От 3-5 години детето започва да изостава от връстниците си във физическото си развитие, като същевременно става апатично, безразлично към хората и събитията.. Типичните знаци са:
- увреждане на речта;
- грапавост на характеристиките;
- редовно незадържане на урина (не само нощно) и изпражнения.
Когато детето порасне, умствената изостаналост се увеличава и в крайна сметка става основен симптом на този вид мукополизахаридоза..
Физическият преглед показва:
- незначително увеличение на черния дроб и далака;
- равномерно увеличаване на разпределението на косата;
- контрактура;
- забавяне на растежа.
 Нарушения на органа на зрението и сърдечно-съдовата система, които често се срещат при други форми на описаната патология, за този вид мукополизахаридоза не са типични.
Нарушения на органа на зрението и сърдечно-съдовата система, които често се срещат при други форми на описаната патология, за този вид мукополизахаридоза не са типични.
Резултатите от рентгеновото изследване с описаната патология или без патологични промени, или подобни на тези, които са открити при мукополизахаридоза тип I-S, но по-слабо изразени.
Прогнозата за това заболяване е по-неблагоприятна, отколкото при други видове мукополизахароза - такива пациенти умират на възраст от 10 до 20 години, инфекциозните усложнения стават причина за смъртта..
Мукополизахаридоза тип IV
Мукополизахаридоза тип IV се нарича също болест на Morkio (кръстен на уругвайския педиатър, който го описва). Патологията е по-често от другите видове мукополизахаридоза, а именно при 1 дете за 40 хиляди новородени.
Деца с това заболяване до 1-3 години се развиват без особености. След това се появяват следните нарушения:
- детето значително изостава от връстниците си;
- в процеса на развитие той отбелязва скъсяване на шията и торса;
- развиват се различни контрактури;
- кожата се сгъстява;
- намалява мускулната сила;
- слухът се влошава;
- чертите на лицето стават груби;
- възникват различни деформации на гърдите.
Също за мукополизахаридоза тип IV има нарушения, които могат да се разглеждат като отделни патологии:
- валгусова деформация на крайниците - кривина в посока навън;
- сколиоза (изкривяване на гръбначния стълб в страничната равнина) или кифоза;
- ингвинална херния;
- пъпна херния;
- дистрофия на роговицата.
Интелект с тази форма на мукополизахаридоза спасен.
Диагнозата на описаната патология се потвърждава с рентгенова снимка, в този случай тя е най-информативна. Това са следните типове:
- Рентгенова снимка на гръбначния стълб - изображенията показват кривината на гръбначния стълб под формата на кифоза и сколиоза, както и разширяването и изправянето на гръбначните тела;
- рентгенография на таза - разкрива многобройни деформации на пръстена на тазовата кост, неравностите на неговите контури;
- Откриват се рентгенография на крайниците - изравняване на главите на бедрената кост, изразено скъсяване на костите на предмишницата и се откриват деформации на костите на крака..
Пациентите с мукополизахаридоза тип IV живеят като правило по-малко от 20 години. Смъртен изход се случва на фона на редица свързани заболявания, които в крайна сметка се усложняват от кардиопулмонарна недостатъчност..
Мукополизахаридоза тип VI
Друго име за мукополизахаридоза тип VI е болестта на Марото-Лами (след имената на двама френски лекари, които описват тази патология)..
Клиничните признаци до голяма степен са "отразени" със симптомите на други видове мукополизахаридоза, но най-силно изразени са:
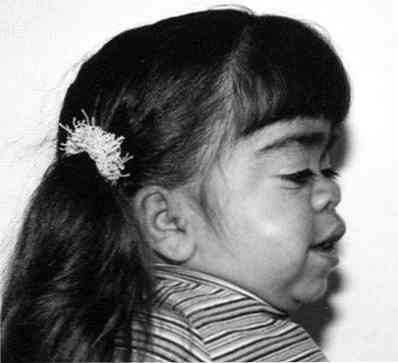 значително нарастване на чертите на лицето;
значително нарастване на чертите на лицето;- забележимо забавяне на растежа;
- скъсяване на шията - понякога изглежда, че главата на пациента "седи" почти на раменния си пояс;
- деформация на гръдния кош.
 значително нарастване на чертите на лицето;
значително нарастване на чертите на лицето;Такива пациенти много често имат настинки - този симптом дава възможност да се подозира това, а не друга форма на мукополизахаридоза..
Физическият преглед показва следното:
- черният дроб и далака често се увеличават;
- може да се диагностицира ингвинална херния.
Интелектуално, детето се развива без нарушения - той има способността да мисли и решава интелектуални проблеми..
Има два варианта на този вид мукополизахаридоза:
- класически;
- мукополизахаридоза с малки клинични прояви.
За да потвърдите диагнозата ще помогне рентгеновото изследване - изображенията се определят от:
- деформация на прешлените - те стават кубовидни или клиновидни;
- триъгълна деформация на тазовия пръстен;
- хипоплазия (недоразвитие) и деформация на главите на бедрената кост;
- скъсяване на фибуловите кости.
Мукополизахаридози тип VII и VIII
 Мукополизахаридозата от тези видове е по-рядко срещана от другите видове описана патология..
Мукополизахаридозата от тези видове е по-рядко срещана от другите видове описана патология..
Мукополизахаридоза тип VII клинично се среща като заболяване от тип III, разликата между тях се открива само в резултатите от биохимичните проучвания..
Мукополизахаридоза тип VIII е симптоматична и подобна на мукополизахаридоза тип IV. Но има разлика в проявите - това е нарушение на интелектуалното развитие, което не се наблюдава при мукополизахаридоза тип IV.
диагностика
При поставяне на диагноза мукополизахаридоза, те се основават на оплаквания, анамнестични данни, резултати от допълнителни методи за изследване - физически, инструментални, лабораторни. Човек може да подозира наличието на мукополизахаридоза от характерните деформации на скелета, които "хващат окото", но е възможно да се определи вида на описаното заболяване само с помощта на допълнителни методи за изследване.. Основните методи са:
- Рентгенови лъчи;
- откриване на гликозаминогликани в урината;
- определяне на ензимната активност в клетъчни култури.
При клинични признаци на нарушения на вътрешните органи се включват методи на изследване:
- електрокардиография;
- ултразвук на черния дроб и далака;
- електроенцефалография;
- биохимичен кръвен тест
и много други.
Диференциална диагностика
Диференциална (отличителна) диагноза се извършва между различните видове мукополизахаридоза.
усложнения
Най-честите усложнения на мукополизахаридозата са:
- контрактура;
- сърдечна недостатъчност;
- белодробна недостатъчност.
лечение
Не е разработена терапия за мукополизахаридоза, която може да се използва за елиминиране на заболяването, а не само за неговите прояви.. Такива пациенти получават симптоматично лечение - може да бъде:
- консервативно - кръвопреливане, хормонална терапия, витаминна терапия и т.н .;
- оперативно - ендопротезиране на главите на бедрената кост, херния, хирургическа интервенция за отстраняване на контрактурите и коригиране на скелетните деформации и др..
Важни са:
- лечение на инфекции на дихателните пътища;
- корекция на зрителното и слуховото увреждане.
предотвратяване
Тъй като патологията възниква поради генетични заболявания, няма специфични превантивни методи.. Но рискът от генни нарушения, които могат да доведат до развитието на различни видове мукополизахаридоза, може да бъде намален, ако жената е снабдена с нормалните условия на бременност. Това е:
- отхвърляне на лоши навици;
- придържане към работа, почивка, сън;
- оптимистично настроение.
перспектива

Прогнозата за мукополизахаридоза като цяло е неблагоприятна, това се отнася за всички видове описани заболявания. В процеса на живот и развитие в тъканите прогресивно се натрупват метаболитни продукти, които възникват поради липса на тъканни ензими. Това води до патологични промени от страна на почти всички органи и системи. Ако патологията започне, смъртта може да настъпи не само поради кардиопулмонална, но и полисистемна недостатъчност.
Консервативната терапия играе ролята на подкрепа - употребата на каквито и да е медикаменти води до временно подобрение, но не може да компенсира липсата на тъканни ензими..
Ковтонюк Оксана Владимировна, медицински коментатор, хирург, консултант